Prevalence of aromatic L-amino acid decarboxylase deficiency in at-risk populations
Hyland K, Reott M. Ped Neurol. 2020;106:38–42.
Publication Date | May 2020
Authors | Hyland K, Reott M.
Citation | Ped Neurol. 2020;106:38–42. doi:10.1016/j.pediatrneurol.2019.11.022
https://pubmed.ncbi.nlm.nih.gov/32111562/
Researchers in Atlanta, US, retrospectively analysed data obtained from samples submitted to Medical Neurogenetics Laboratories, a US-based clinical diagnostics company, to estimate the prevalence of aromatic L-amino acid decarboxylase (AADC) deficiency in an at-risk population.1
AADC deficiency is a rare, autosomal recessive metabolic disorder in which pathogenic mutations in the dopa decarboxylase (DDC) gene result in reduced AADC enzyme activity and severe combined deficiencies in dopamine, serotonin, norepinephrine and epinephrine production. This leads to a complex syndrome characterised by motor, behavioural and autonomic symptoms.2 There are around 120 confirmed diagnoses of AADC deficiency worldwide documented in the literature. A founder mutation (IVS6+4A>T) has been identified in East Asia, with the prevalence of AADC deficiency in Taiwan estimated to be 1:32,000, however, little is known about the prevalence of AADC deficiency elsewhere.1
In the 68 cases documented in the literature, the average age of symptom onset for AADC deficiency is 2.7 months. Despite early onset of symptoms, the average age of diagnosis is 3.5 years. The 2017 Consensus Guidelines for Diagnosis of AADC deficiency require at least 2 out of 3 core criteria to be met for a diagnosis of AADC deficiency: (1) low cerebrospinal fluid (CSF) levels of 5-hydroxyindoleacetic acid (5-HIAA), homovanillic acid (HVA), and 3-methoxy-4-hydroxyphenylglycol with increased CSF levels of 3-O-methyldopa (3-OMD), levodopa (L-Dopa), and 5-hydroxytryptophan (5-HTP), and normal CSF pterins; (2) decreased AADC enzyme activity in plasma; and (3) compound heterozygous or homozygous pathologic variants in the DDC gene.2
In this retrospective study,1 investigators Keith Hyland and Michael Reott from the Department of Neurochemistry, Medical Neurogenetics Laboratories, examined the diagnostic data from almost 20,000 samples from at-risk patients presenting with neurological deficits of unknown origin. Samples were submitted from around the world between 2008 and 2016 for analysis of neurotransmitter metabolites in CSF, AADC enzyme activity in plasma, and/or Sanger sequencing of the DDC gene.
Overall, 36 new cases of AADC deficiency were identified from 19,684 unique samples. 19,577 of these were CSF samples tested for neurotransmitter metabolite levels and 22 patients with AADC deficiency were identified, resulting in an estimated prevalence of 0.112% (1:900). CSF analysis alone was used for diagnosis of 3 of these patients; 7 had their diagnosis further confirmed through assessment of plasma AADC enzyme activity, and 3 AADC deficiency cases were confirmed with Sanger sequencing of the DDC gene. Both of these additional analyses were used as confirmation for 9 patients. Sanger sequencing alone identified 5 patients with AADC deficiency, plasma enzyme activity assessment alone identified 4 patients, and both of these tests were used to confirm a diagnosis of 5 further patients (Figure 1).
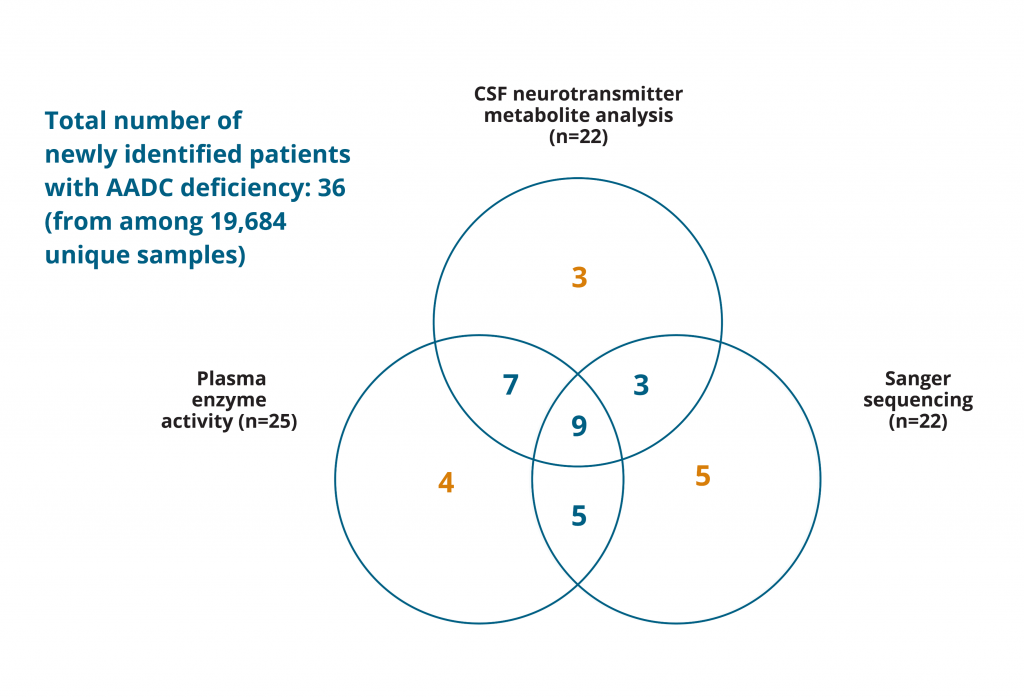
Adapted from: Hyland K, Reott M. Ped Neurol. 2020;106:38–42.
Figure 1: Retrospective examination of AADC deficiency cases in an at-risk population. Numbers in orange indicate cases identified as AADC deficiency according to retrospectively analysed data with only 1 of the diagnostic criteria being met;1 numbers in blue indicate cases diagnosed as AADC deficiency based on the 2017 Consensus Guidelines diagnostic criteria of any 2 of the core tests being positive.2
In total, the retrospective analysis identified 36 new cases of AADC deficiency, with a median age at diagnosis of approximately 2.75 years. However, if only the cases that conformed to the consensus guidelines — i.e. meeting at least 2 of the 3 diagnostic criteria — were included in the retrospective analysis, it was estimated that there would have been 24 newly identified cases, a prevalence of 0.097%, or roughly 1:1,000.
The authors stated that, in their own experience, any one of the three diagnostic criteria should be sufficient for a diagnosis of AADC deficiency without the need for any accompanying analysis.
The DNA sequencing results identified 26 different variants of the DDC gene, including the founder mutation IVS6+4A>T, which was identified in 8 patients, one of whom was homozygous for this change. Another variant, p.(Arg347Gln), was identified in 7 alleles, which the authors suggested could be indicative of another founder mutation. Five of the variants identified had not previously been associated with AADC deficiency but were deemed to be likely pathogenic due to the knowledge of the plasma enzyme activity and CSF metabolite levels, illustrating the importance of functional studies when investigating the pathogenicity of variant changes in the DDC gene.
The results of this study add to the growing evidence base surrounding the diagnosis and management of AADC deficiency, supporting the current understanding of the epidemiology and genetics of the condition, as well as adding to the expanding list of known pathogenic variants of the DDC gene.
- Hyland K, Reott M. Ped Neurol. 2020;106:38–42.
- Wassenberg T, et al. Orphanet J Rare Dis. 2017;12:12.